Sterile drugs should be manufactured using aseptic processing only when terminal sterilization is not feasible. However, some final packaging may afford some unique and substantial advantage (e.g., some dual-chamber syringes) that would not be possible if terminal sterilization were employed. In such cases, a manufacturer can explore the option of adding adjunct processing steps to increase the level of sterility assurance.
Product Requirements
FDA has stated, where possible, that the use of terminal sterilization should be used in the manufacture of sterile product, when no detrimental effects has been inflicted on the achieving the end-product.
Consequently, the first step in establishing the processing conditions, and therefore the design of the manufacturing facility, is to determine whether terminal sterilization will be requires.
In some cases, an additional method e.g. heat treatment, can be added to aseptic processing to better assure product safety.
Decision Tree for the Selection of the Sterilization Methods
- Those products intended to be sterile should be terminally sterilized in their final container as clearly stated in the European Pharmacopoeia, and in the CPMP Notes for Guidance.
- Where it is not possible to carry out terminal sterilization by heat due to formulation instability, a decision should be taken to utilize an alternative method of terminal sterilization, filtration and/or aseptic processing.
- It is recognized that the new terminal sterilization process other than those described in the pharmacopoeia may be developed to provide sterility assurance levels equivalent to present official methods, and such processes when properly validated may offer alternative approaches.
- The use of an inappropriate heat-labile packaging material cannot in itself be the sole reason for adoption of aseptic processing. However, it may be that the choice of a packaging material for a given product has to take into account factors other than the method of sterilization.
- Conventionally, it has been accepted that other factors such as the type of container, route of administration and patient benefit have contributed to the choice of a particular container type, which will not withstand terminal heat sterilization (e.g. certain ophthalmic products) and such products are therefore manufactured by validated aseptic processing.
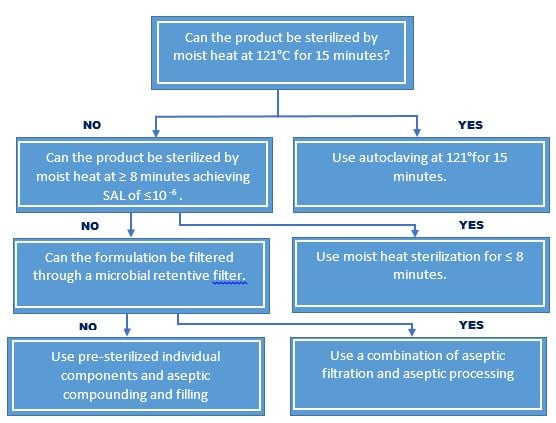
Sterilization Choices for Aqueous Products
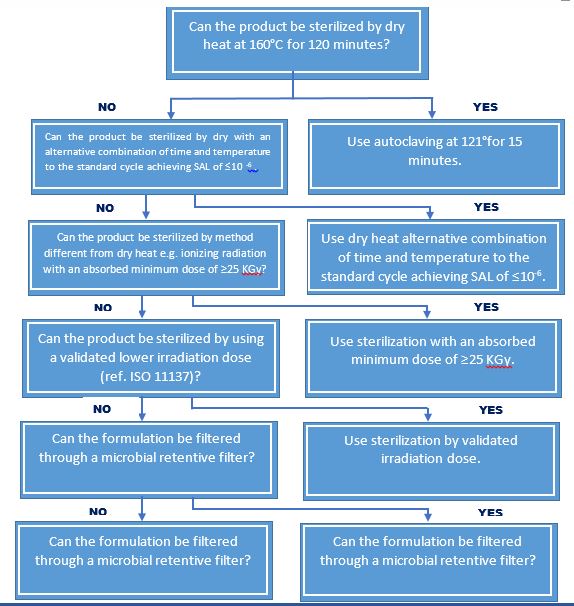
Sterilization Choices for Non Aqueous Liquid, Semi solid, or Dry Powder
For the purpose of ensuring sterility, all aqueous-based sterile products are subject to terminal moist heat sterilization, with the following exceptions:
- Instances where terminal moist heat sterilization is not practical, e.g. product degradation. Such instances are fully evaluated and documented.
- For aseptic processes that exclude human intervention e.g., robotics, form-fill-seal and barrier system, may be employed in lieu of terminal moist heat sterilization providing that validation data demonstrated equivalence.
Two basic approaches are employed to develop sterilization cycles for moist heat processes:
- Overkill
This is used when the product can withstand excessive heat treatment such as an F0 ≥ 12 without adverse effects. Bio burden and resistance data are not required to determine the required “F0“values. Cycle parameters are adjusted to assure that the coldest point within the load receives an “F0“that will provide at least a 12-log reduction of microorganisms having a D121 value of at least one minute.
- Probability of survival
This approach is used primarily for heat labile products. In this approach, the process for the terminal sterilization of a sealed container is validated to achieve the destruction of pre-sterilization bio burden to a level of 100, with a minimum safety factor of an additional six-log reduction (1x10-6). The probability that any one unit is contaminated is therefore no more than one in a million; this is considered to be an acceptable level of sterility assurance.
The probability of survival is determined using a semi-logarithmic microbial death curve, where a plot of the log of the number of survivor versus time at a fixed temperature yields a straight line.
The determination of the minimum “F0“value for the probability of survival approach is based upon the number of microorganisms (bio burden) found in a given product and their heat resistance.
Types of Terminal Sterilization
Sterilization by heat
Each heat-sterilization cycle should be recorded by means of appropriate equipment of suitable accuracy and precision, e.g. on a time/temperature chart with a suitably large scale. The temperature should be recorded by a probe at the coolest part of the load or loaded chamber, this point having been determined during the validation; the temperature should preferably be checked against a second independent temperature probe located at the same position. The chart, or a photocopy of it, should form part of the batch record. Chemical or biological indicators may also be used but should not take the place of physical controls.
Sufficient time must be allowed for the whole of the load to reach the required temperature before measurement of the sterilizing time is started. This time must be determined for each type of load to be processed.
After the high-temperature phase of a heat sterilization cycle, precautions should be taken against contamination of a sterilized load during cooling. Any cooling fluid or gas in contact with the product should be sterilized.
Sterilization by moist heat
Sterilization by moist heat (heating in an autoclave) is suitable only for water-wettable materials and aqueous formulations. Both temperature and pressure should be used to monitor the process. The temperature recorder should normally be independent of the controller, and there should be an independent temperature indicator, the reading from which should be routinely checked against the chart recorder during the sterilization period. For sterilizers fitted with a drain at the bottom of the chamber, it may also be necessary to record the temperature at this position throughout the sterilization period. There should be regular leak tests on the chamber when a vacuum phase is part of the cycle.
The items to be sterilized, other than products in sealed containers, should be wrapped in a material that allows the removal of air and the penetration of steam but prevents recontamination after sterilization. All parts of the load should be in contact with water or saturated steam at the required temperature for the required time.
Care should be taken to ensure that the steam used for sterilization is of suitable quality and does not contain additives at a level that could cause contamination of the product or equipment.
Sterilization by dry heat
Sterilization by dry heat may be suitable for non-aqueous liquids or dry powder products. The process used should include air circulation within the chamber and the maintenance of a positive pressure to prevent the entry of non-sterile air. If air is supplied, it should be passed through a microorganism-retaining filter (e.g. an HEPA filter). Where sterilization by dry heat is also intended to remove pyrogens, challenge tests using endotoxins will be required as part of the validation.
Sterilization by radiation
Sterilization by radiation is used mainly for the sterilization of heat-sensitive materials and products. Many pharmaceutical products and some packaging materials are radiation-sensitive, so this method is permissible only when the absence of deleterious effects on the product has been confirmed experimentally. Ultraviolet irradiation is not an acceptable method for terminal sterilization.
If sterilization by radiation is carried out by an outside contractor, the manufacturer is responsible for ensuring that the requirements of section 6.8 are met, and that the sterilization process is validated. The responsibilities of the radiation plant operator (e.g. for using the correct dose) should also be specified.
During the sterilization procedure, the radiation dose should be measured. For this purpose, the dosimeters used must be independent of the dose rate and must provide a quantitative measurement of the dose received by the product itself. Dosimeters should be inserted in the load in sufficient number, and close enough together to ensure that there is always a dosimeter in the chamber. Where plastic dosimeters are employed, they should be used within the time-limit of their calibration. Dosimeter absorbances should be read shortly after exposure to radiation. Biological indicators may be used only as an additional control. Radiation-sensitive colour discs may be used to differentiate between packages that have been subjected to irradiation and those that have not; they are not indicators of successful sterilization. The information obtained should constitute part of the batch record.
Validation procedures should ensure that consideration is given to the effects of variations in the density of the packages.
Handling procedures should prevent any misidentification of irradiated and non-irradiated materials. Each package should carry a radiation-sensitive indicator to show whether or not it has been subjected to radiation treatment.
The total radiation dose should be administered within a predetermined period of time.
Sterilization by gases and fumigants
This method of sterilization should only be used for products where there is no suitable alternative.
Various gases and fumigants may be used for sterilization (e.g. ethylene oxide, hydrogen peroxide vapour). Ethylene oxide should be used only when no other method is practicable. During process validation it should be shown that the gas has no damaging effect on the product and that the conditions and time allowed for degassing are such as to reduce any residual gas and reaction products to defined acceptable limits for the type of product or material concerned. These limits should be incorporated in the specifications.
Direct contact between gas and microorganisms is essential; precautions should therefore be taken to avoid the presence of organisms likely to be enclosed in materials such as crystals or dried protein. The nature and quantity of packaging materials can significantly affect the process.
Before exposure to the gas, materials should be brought into equilibrium with the humidity and temperature required by the process. This requirement should be balanced against the need to minimize the waiting time before sterilization.
Each sterilization cycle should be monitored with suitable biological indicators, using the appropriate number of test pieces distributed throughout the load. The information so obtained should form part of the batch record.
Biological indicators should be stored and used according to the manufacturer’s instructions, and their performance checked by positive controls.
For each sterilization cycle, records should be made of the time taken to complete the cycle, of the pressure, temperature and humidity within the chamber during the process, and of the gas concentration. The pressure and temperature should be recorded on a chart throughout the cycle. The records should form part of the batch record.
After sterilization, the load should be stored in a controlled manner under ventilated conditions to allow concentrations of residual gas and reaction products to fall to their prescribed levels. This process should be validated.




